Umm… thanks for the GPT… I mad it.
import os
from mp_api.client import MPRester
with MPRester("My_API") as mpr:
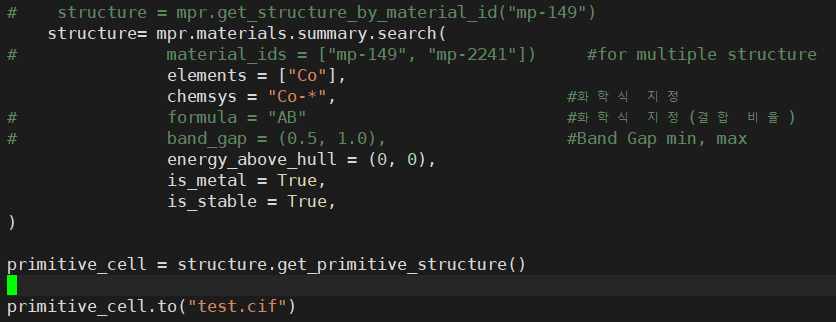
# structure = mpr.get_structure_by_material_id("mp-149")
structures = mpr.materials.summary.search(
# material_ids = ["mp-149""mp-2241"]) #for multiple structure
elements = ["Co"],
chemsys = "Co-*",
# formula = "AB"
# band_gap = (0.51.0),
energy_above_hull = (00),
is_metal = True,
is_stable = True
)
with open("test.txt""w") as file:
file.write(f"Number of documents: {len(structures)}\n\n")
if not structures:
file.write("No results found.\n")
else:
for idxstructure_summary in enumerate(structures):
material_id = structure_summary.material_id
formula = structure_summary.formula_pretty
# E_Hull = structure_summary.energy_above_hull
# Is_Metal = structure_summary.is_metal
# stable = structure_summary.is_stable
# crystal = structure_summary.structure
sym = structure_summary.symmetry
structure = mpr.get_structure_by_material_id(material_id)
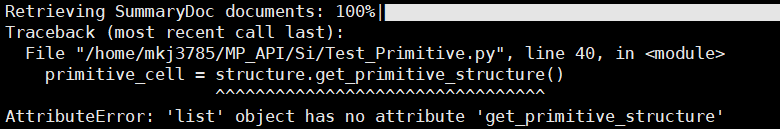
primitive_cell = structure.get_primitive_structure()
cif_filename = f"{formula}.cif"
primitive_cell.to(filename = cif_filename)
file.write(f"--- Document {idx + 1} ---\n")
file.write(f"Material ID: {material_id}\n")
# file.write(f"Chemical Formula: {formula}\n")
# file.write(f"Energy Above Hull: {E_Hull}\n")
# file.write(f"Is Metal: {Is_Metal}\n")
# file.write(f"Stable: {stable}\n")
# file.write(f"Crystal: {structure}\n")
file.write(f"Symmetry: {sym}\n")
file.write(f"Primitive Structure:\n{primitive_cell}\n")
file.write('-' * 40 + "\n")
file.write("\n\n")
First of allI’ve written it this waybut if there are any good options for optimizationplease let me know.
And there is another question.
There is another option to figure out the cell data.
crystal = structure_summary.structure
When I use this optionthe variable “crystal” include whole materials data.
It seems that the structure_summary.structure is same as structure.get_primitive_structure().
Can you tell me if I right?
for idxstructure_summary in enumerate(structures):
material_id = structure_summary.material_id
formula = structure_summary.formula_pretty
sym = structure_summary.symmetry
structure = mpr.get_structure_by_material_id(material_id)
primitive_cell = structure.get_primitive_structure()
file.write(f"--- Document {idx + 1} ---\n")
file.write(f"Material ID: {material_id}\n")
file.write(f"Symmetry: {sym}\n")
file.write(f"Primitive Structure:\n{primitive_cell}\n")
file.write('-' * 40 + "\n")
file.write("\n\n")
output by `structure.get_primitive_structure()`
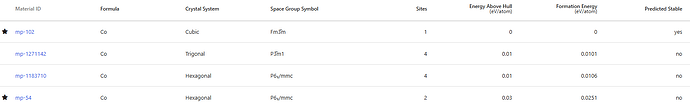
Number of documents: 110
— Document 1 —
Material ID: mp-9945
Symmetry: crystal_system=<CrystalSystem.ortho: ‘Orthorhombic’> symbol=‘Pnnm’ number=58 point_group=‘mmm’ symprec=0.1 version=‘2.0.2’
Primitive Structure:
Full Formula (Co2 Te4)
Reduced Formula: CoTe2
abc : 3.936121 5.322096 6.390892
angles: 90.001887 90.000000 90.000000
pbc : True True True
Sites (6)
SP a b c magmom
0 Co -0 -0 0 1.161
1 Co 0.5 0.5 0.5 1.16
2 Te -0 0.21977 0.365541 -0.052
3 Te -0 0.78023 0.634459 -0.052
4 Te 0.5 0.719763 0.134487 -0.052
5 Te 0.5 0.280237 0.865513 -0.052
— Document 2 —
Material ID: mp-284
Symmetry: crystal_system=<CrystalSystem.cubic: ‘Cubic’> symbol=‘Pm-3m’ number=221 point_group=‘m-3m’ symprec=0.1 version=‘2.0.2’
Primitive Structure:
Full Formula (Al1 Co1)
Reduced Formula: AlCo
abc : 2.819403 2.819403 2.819403
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (2)
SP a b c magmom
0 Al -0 -0 -0 0
1 Co 0.5 0.5 0.5 -0
for idxstructure_summary in enumerate(structures):
material_id = structure_summary.material_id
sym = structure_summary.symmetry
crystal = structure_summary.structure
file.write(f"--- Document {idx + 1} ---\n")
file.write(f"Material ID: {material_id}\n")
file.write(f"Symmetry: {sym}\n")
file.write(f"Crystal: {crystal}\n")
file.write('-' * 40 + "\n")
file.write("\n\n")
output by `structure_summary.structure`
Number of documents: 110
— Document 1 —
Material ID: mp-9945
Symmetry: crystal_system=<CrystalSystem.ortho: ‘Orthorhombic’> symbol=‘Pnnm’ number=58 point_group=‘mmm’ symprec=0.1 version=‘2.0.2’
Crystal: Full Formula (Co2 Te4)
Reduced Formula: CoTe2
abc : 3.936121 5.322096 6.390892
angles: 90.001887 90.000000 90.000000
pbc : True True True
Sites (6)
SP a b c magmom
0 Co -0 -0 0 1.161
1 Co 0.5 0.5 0.5 1.16
2 Te -0 0.21977 0.365541 -0.052
3 Te -0 0.78023 0.634459 -0.052
4 Te 0.5 0.719763 0.134487 -0.052
5 Te 0.5 0.280237 0.865513 -0.052
— Document 2 —
Material ID: mp-284
Symmetry: crystal_system=<CrystalSystem.cubic: ‘Cubic’> symbol=‘Pm-3m’ number=221 point_group=‘m-3m’ symprec=0.1 version=‘2.0.2’
Crystal: Full Formula (Al1 Co1)
Reduced Formula: AlCo
abc : 2.819403 2.819403 2.819403
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (2)
SP a b c magmom
0 Al -0 -0 -0 0
1 Co 0.5 0.5 0.5 -0